AMPtk Commands¶
A description for all AMPtk commands.
AMPtk wrapper script¶
AMPtk is a series of Python scripts that are launched from a Python wrapper script. Each command has a help menu which you can print to the terminal by issuing the command without any arguments, i.e. amptk yields the following.
$ amptk
Usage: amptk <command> <arguments>
version: 1.3.0
Description: AMPtk is a package of scripts to process NGS amplicon data.
Dependencies: USEARCH v9.1.13 and VSEARCH v2.2.0
Process: ion pre-process Ion Torrent data
illumina pre-process folder of de-multiplexed Illumina data
illumina2 pre-process PE Illumina data from a single file
illumina3 pre-process PE Illumina + index reads (i.e. EMP protocol)
454 pre-process Roche 454 (pyrosequencing) data
SRA pre-process single FASTQ per sample data (i.e. SRA data)
Clustering: cluster cluster OTUs (using UPARSE algorithm)
dada2 dada2 denoising algorithm (requires R, dada2, ShortRead)
unoise2 UNOISE2 denoising algorithm
unoise3 UNOISE3 denoising algorithm
cluster_ref closed/open reference based clustering (EXPERIMENTAL)
Utilities: filter OTU table filtering
lulu LULU amplicon curation of OTU table
taxonomy Assign taxonomy to OTUs
show show number or reads per barcode from de-multiplexed data
select select reads (samples) from de-multiplexed data
remove remove reads (samples) from de-multiplexed data
sample sub-sample (rarify) de-multiplexed reads per sample
drop Drop OTUs from dataset
stats Hypothesis test and NMDS graphs (EXPERIMENTAL)
summarize Summarize Taxonomy (create OTU-like tables and/or stacked bar graphs)
funguild Run FUNGuild (annotate OTUs with ecological information)
meta pivot OTU table and append to meta data
heatmap Create heatmap from OTU table
SRA-submit De-multiplex data and create meta data for NCBI SRA submission
Setup: install Download/install pre-formatted taxonomy DB. Only need to run once.
database Format Reference Databases for Taxonomy
info List software version and installed databases
primers List primers hard-coded in AMPtk. Can use in pre-processing steps.
version List version
citation List citation
AMPtk Pre-Processing¶
amptk ion¶
Script that demulitplexes Ion Torrent data. Input can be either an unaligned BAM file or FASTQ file. The IonXpress 1-96 barcodes are hard-coded into AMPtk and is the default setting for providing barcode sequences to the script. Alternatively, you can provide a --barcode_fasta file containing barcodes used or a QIIME like mapping file. For file formats see here, and for more information see here.
Usage: amptk ion <arguments>
version: 1.3.0
Description: Script processes Ion Torrent PGM data for AMPtk clustering. The input to this script
should be a FASTQ file obtained from the Torrent Server analyzed with the
`--disable-all-filters` flag to the BaseCaller. This script does the following:
1) finds Ion barcode sequences, 2) relabels headers with appropriate barcode name,
3) removes primer sequences, 4) trim/pad reads to a set length.
Arguments: -i, --fastq,--bam Input BAM or FASTQ file (Required)
-o, --out Output base name. Default: out
-m, --mapping_file QIIME-like mapping file
-f, --fwd_primer Forward primer sequence. Default: fITS7
-r, --rev_primer Reverse primer sequence Default: ITS4
-b, --barcodes Barcodes used (list, e.g: 1,3,4,5,20). Default: all
-n, --name_prefix Prefix for re-naming reads. Default: R_
-l, --trim_len Length to trim/pad reads. Default: 300
-p, --pad Pad reads with Ns if shorter than --trim_len. Default: off [on,off]
--min_len Minimum length read to keep. Default: 100
--full_length Keep only full length sequences.
--barcode_fasta FASTA file containing barcodes. Default: pgm_barcodes.fa
--barcode_mismatch Number of mismatches in barcode to allow. Default: 0
--primer_mismatch Number of mismatches in primers to allow. Default: 2
--cpus Number of CPUs to use. Default: all
--mult_samples Combine multiple chip runs, name prefix for chip
amptk illumina¶
Script for demultiplexing Illumina PE data that has been delivered from sequencing center in a folder of PE FASTQ files, one set for each sample. More information is here.
Usage: amptk illumina <arguments>
version: 1.3.0
Description: Script takes a folder of Illumina MiSeq data that is already de-multiplexed
and processes it for clustering using AMPtk. The default behavior is to:
1) merge the PE reads using USEARCH, 2) find and trim primers, 3) rename reads
according to sample name, 4) trim/pad reads to a set length.
Arguments: -i, --fastq Input folder of FASTQ files (Required)
-o, --out Output folder name. Default: amptk-data
-m, --mapping_file QIIME-like mapping file
-f, --fwd_primer Forward primer sequence. Default: fITS7
-r, --rev_primer Reverse primer sequence Default: ITS4
-l, --trim_len Length to trim/pad reads. Default: 300
-p, --pad Pad reads with Ns if shorter than --trim_len. Default: off [on,off]
--min_len Minimum length read to keep. Default: 100
--full_length Keep only full length sequences.
--reads Paired-end or forward reads. Default: paired [paired, forward]
--read_length Illumina Read length (250 if 2 x 250 bp run). Default: auto detect
--rescue_forward Rescue Forward Reads if PE do not merge, e.g. long amplicons. Default: on [on,off]
--require_primer Require the Forward primer to be present. Default: on [on,off]
--primer_mismatch Number of mismatches in primers to allow. Default: 2
--barcode_mismatch Number of mismatches in barcode to allow. Default: 1
--cpus Number of CPUs to use. Default: all
--cleanup Remove intermediate files.
--merge_method Software to use for PE merging. Default: usearch [usearch,vsearch]
-u, --usearch USEARCH executable. Default: usearch9
amptk illumina2¶
This script is for demultiplexing Illumina data that is delivered as either a single FASTQ file or PE FASTQ files where the read layout contains unique barcode sequences at the 5’ or the 3’ end of the amplicons. More information is here.
Usage: amptk illumina2 <arguments>
version: 1.3.0
Description: Script takes Illumina data that is not de-multiplexed and has read structure
similar to Ion/454 such that the reads are <barcode><fwd_primer>Read<rev_primer> for
clustering using AMPtk. The default behavior is to: 1) find barcodes/primers,
2) relabel headers and trim barcodes/primers, 3) merge the PE reads,
4) trim/pad reads to a set length. This script can handle dual barcodes
(3' barcodes using the --reverse_barcode option or mapping file).
Arguments: -i, --fastq Illumina R1 (PE forward) reads (Required)
--reverse Illumina R2 (PE reverse) reads.
-o, --out Output base name. Default: illumina2
-m, --mapping_file QIIME-like mapping file
-f, --fwd_primer Forward primer sequence. Default: fITS7
-r, --rev_primer Reverse primer sequence Default: ITS4
-n, --name_prefix Prefix for re-naming reads. Default: R_
-l, --trim_len Length to trim/pad reads. Default: 300
-p, --pad Pad reads with Ns if shorter than --trim_len. Default: off [on,off]
--min_len Minimum length read to keep. Default: 100
--barcode_fasta FASTA file containing barcodes.
--reverse_barcode FASTA file containing R2 barcodes.
--barcode_mismatch Number of mismatches in barcode to allow. Default: 0
--barcode_not_anchored Barcodes are not anchored to start of read.
--full_length Keep only full length sequences.
--primer_mismatch Number of mismatches in primers to allow. Default: 2
--merge_method Software to use for PE merging. Default: usearch [usearch,vsearch]
--cpus Number of CPUs to use. Default: all
-u, --usearch USEARCH executable. Default: usearch9
amptk illumina3¶
This script demultiplexes Illumina PE data that is delivered as 3 files: forward reads (R1), reverse reads (R2), and then index reads (I3). More information is here.
Usage: amptk illumina3/emp <arguments>
version: 1.3.0
Description: Script takes PE Illumina reads, Index reads, mapping file and processes
data for clustering/denoising in AMPtk. The default behavior is to:
1) find and trim primers, 2) merge the PE reads, 3) filter for Phix,
4) rename reads according to sample name, 4) trim/pad reads.
Arguments: -f, --forward FASTQ R1 (forward) file (Required)
-r, --reverse FASTQ R2 (reverse) file (Required)
-i, --index FASTQ I3 (index) file (Required)
-m, --mapping_file QIIME-like mapping file.
-l, --trim_len Length to trim/pad reads. Default: 300
-p, --pad Pad reads with Ns if shorter than --trim_len. Default: off [on,off]
-o, --out Output folder name. Default: amptk-data
--fwd_primer Forward primer sequence
--rev_primer Reverse primer sequence
--min_len Minimum length read to keep. Default: 100
--read_length Illumina Read length (250 if 2 x 250 bp run). Default: auto detect
--rescue_forward Rescue Forward Reads if PE do not merge, e.g. long amplicons. Default: on [on,off]
--barcode_fasta Multi-fasta file of barocdes.
--primer_mismatch Number of mismatches in primers to allow. Default: 2
--barcode_mismatch Number of mismatches in index (barcodes) to allow. Default: 2
--barcode_rev_comp Reverse complement barcode sequences in mapping file.
--merge_method Software to use for PE merging. Default: usearch [usearch,vsearch]
--cpus Number of CPUs to use. Default: all
--cleanup Remove intermediate files.
-u, --usearch USEARCH executable. Default: usearch9
amptk 454¶
Script for demultiplexing Roche 454 data. Input requirements are a 454 run in SFF, FASTQ, or FASTA+QUAL format as well as a multi-FASTA file containing barcodes used. More information is here.
Usage: amptk 454 <arguments>
version: 1.3.0
Description: Script processes Roche 454 data for AMPtk clustering. The input to this script
should be either a SFF file, FASTA+QUAL files, or FASTQ file. This script does
the following: 1) finds barcode sequences, 2) relabels headers with appropriate
barcode name, 3) removes primer sequences, 4) trim/pad reads to a set length.
Arguments: -i, --sff, --fasta Input file (SFF, FASTA, or FASTQ) (Required)
-q, --qual QUAL file (Required if -i is FASTA).
-o, --out Output base name. Default: out
-m, --mapping_file QIIME-like mapping file
-f, --fwd_primer Forward primer sequence. Default: fITS7
-r, --rev_primer Reverse primer sequence Default: ITS4
-n, --name_prefix Prefix for re-naming reads. Default: R_
-l, --trim_len Length to trim/pad reads. Default: 250
-p, --pad Pad reads with Ns if shorter than --trim_len. Default: off [on,off]
--min_len Minimum length read to keep. Default: 50
--barcode_fasta FASTA file containing barcodes. (Required)
--reverse_barcode FASTA file containing 3' barcodes. Default: none
--barcode_mismatch Number of mismatches in barcode to allow. Default: 0
--primer_mismatch Number of mismatches in primers to allow. Default: 2
--cpus Number of CPUs to use. Default: all
amptk SRA¶
This script is useful for pre-processing data from the NCBI SRA or data that is located in a folder where each sample is contained in a single FASTQ file. Note if you have PE Illumina data that was downloaded from SRA, you can use the amptk illumina script. More information is here.
Usage: amptk SRA <arguments>
version: 1.3.0
Description: Script takes a folder of FASTQ files in a format you would get from NCBI SRA, i.e.
there is one FASTQ file for each sample. Reads will be named according to sample name
and workflow is 1) find and trim primers, 2) rename reads according to filename,
and 3) trim/pad reads to a set length (optional).
Arguments: -i, --fastq Input folder of FASTQ files (Required)
-o, --out Output folder name. Default: amptk-data
-m, --mapping_file QIIME-like mapping file
-f, --fwd_primer Forward primer sequence. Default: fITS7
-r, --rev_primer Reverse primer sequence Default: ITS4
-l, --trim_len Length to trim/pad reads. Default: 250
-p, --pad Pad reads with Ns if shorter than --trim_len. Default: off [on,off]
--min_len Minimum length read to keep. Default: 50
--full_length Keep only full length sequences.
--require_primer Require the Forward primer to be present. Default: on [on,off]
--primer_mismatch Number of mismatches in primers to allow. Default: 2
--cpus Number of CPUs to use. Default: all
--cleanup Remove intermediate files.
-u, --usearch USEARCH executable. Default: usearch9
AMPtk Clustering¶
amptk cluster¶
UPARSE clustering in AMPtk is completed with this command. There is optional reference based chimera filtering. More information is here.
Usage: amptk cluster <arguments>
version: 1.3.0
Description: Script is a "wrapper" for the UPARSE algorithm. FASTQ quality trimming via expected
errors and dereplication are run in vsearch if installed otherwise defaults to Python
which allows for the use of datasets larger than 4GB.
Chimera filtering and UNOISE are also options.
Arguments: -i, --fastq Input FASTQ file (Required)
-o, --out Output base name. Default: out
-e, --maxee Expected error quality trimming. Default: 1.0
-p, --pct_otu OTU Clustering Radius (percent). Default: 97
-m, --minsize Minimum size to keep (singleton filter). Default: 2
--uchime_ref Run Ref Chimera filtering. Default: off [ITS, LSU, COI, 16S, custom path]
--map_filtered Map quality filtered reads back to OTUs. Default: off
--unoise Run De-noising pre-clustering (UNOISE). Default: off
--debug Keep intermediate files.
--cpus Number of CPUs to use. Default: all
-u, --usearch USEARCH executable. Default: usearch9
amptk dada2¶
DADA2 infers exact sequence variants (ESVs or iSeqs) by using a statistical error model to correct sequencing errors. AMPtk employs a modified DADA2 workflow that also clusters the iSeqs into biological meaningful OTUs. More information is here.
Usage: amptk dada2 <arguments>
version: 1.3.0
Description: Script is a "wrapper" for the DADA2 pipeline. It will "pick OTUs" based on denoising
the data for each read predicting the original sequence. This pipeline is sensitive to
1 bp differences between sequences. Since most reference databases classify "species"
at 97%% threshold, the inferred sequences (iSeqs) from DADA2 are then clusterd at --pct_otu
to create OTUs. Both results are saved. Requires R packages: dada2, ShortRead
Arguments: -i, --fastq Input FASTQ file (Required)
-o, --out Output base name. Default: dada2
-m, --min_reads Minimum number of reads per sample. Default: 10
-l, --length Length to trim reads.
-e, --maxee Expected error quality trimming. Default: 1.0
-p, --pct_otu OTU Clustering Radius (percent). Default: 97
--platform Sequencing platform. [ion, illumina, 454]. Default: ion
--pool Pool all samples together for DADA2. Default: off
--uchime_ref Run Ref Chimera filtering. Default: off [ITS, LSU, COI, 16S, custom path]
--cpus Number of CPUs to use. Default: all
--debug Keep intermediate files.
amptk unoise2¶
UNOISE2 is a denoising algorithm in USEARCH9 that was built to work in a similar fashion to DADA2, correcting reads instead of clustering them. More information is here.
Usage: amptk unoise2 <arguments>
version: 1.3.0
Description: Script will run the UNOISE2 denoising algorithm followed by clustering with
UCLUST to generate OTUs. OTU table is then constructed by mapping reads to
the OTUs. Requires USEARCH v9.0.232 or greater.
Arguments: -i, --fastq Input FASTQ file (Required)
-o, --out Output base name. Default: out
-e, --maxee Expected error quality trimming. Default: 1.0
-m, --minsize Minimum size to keep for denoising. Default: 8
-p, --pct_otu OTU Clustering Radius (percent). Default: 97
-u, --usearch Path to USEARCH9. Default: usearch9
--uchime_ref Run Ref Chimera filtering. Default: off [ITS, LSU, COI, 16S, custom path]
--cpus Number of CPUs to use. Default: all
--debug Keep intermediate files.
amptk unoise3¶
UNOISE3 is the successor to UNOISE2 and is a denoising algorithm built from the Illumina platform. The author suggests that 454 and Ion Torrent data do not work well with this method. More information is here.
Usage: amptk unoise3 <arguments>
version: 1.3.0
Description: Script will run the UNOISE3 denoising algorithm followed by clustering with
UCLUST to generate OTUs. OTU table is then constructed by mapping reads to
the OTUs. Requires USEARCH v10.0.240 or greater.
Arguments: -i, --fastq Input FASTQ file (Required)
-o, --out Output base name. Default: out
-e, --maxee Expected error quality trimming. Default: 1.0
-m, --minsize Minimum size to keep for denoising. Default: 8
-p, --pct_otu OTU Clustering Radius (percent). Default: 97
-u, --usearch Path to USEARCH9. Default: usearch9
--uchime_ref Run Ref Chimera filtering. Default: off [ITS, LSU, COI, 16S, custom path]
--cpus Number of CPUs to use. Default: all
--debug Keep intermediate files.
amptk cluster_ref¶
This script runs reference based clustering or rather maps each unique sequence to a reference database using global alignment. If a sequence has no match greather than --id, the remaining sequences are classified using UTAX.
Usage: amptk cluster_ref <arguments>
version: 1.3.0
Description: Script first quality filters reads, dereplicates, and then runs chimera
filtering. OTUs are then picked via reference based clustering (closed)
those that are > --id. The rest of the data can then be clustered via
de novo UPARSE and then reference clustered using UTAX. EXPERIMENTAL
Arguments: -i, --fastq Input FASTQ file (Required)
-d, --db Database [ITS,ITS1,ITS2,16S,LSU,COI,custom]. (Required)
-o, --out Output base name. Default: out
-e, --maxee Expected error quality trimming. Default: 1.0
-p, --pct_otu OTU Clustering Radius (percent). Default: 97
-m, --minsize Minimum size to keep (singleton filter). Default: 2
--id Percent ID for closed reference clustering. Default: 97
--utax_db UTAX formatted DB.
--utax_level UTAX Taxonomy level to keep. Default: k [k,p,c,o,f,g,s]
--utax_cutoff UTAX confidence value threshold. Default: 0.8 [0 to 0.9]
--mock Mock community fasta file
--closed_ref_only Run only closed reference clustering.
--map_filtered Map quality filtered reads back to OTUs. Default: off
--debug Keep intermediate files.
--cpus Number of CPUs to use. Default: all
-u, --usearch USEARCH executable. Default: usearch9
AMPtk Utilities¶
amptk filter¶
Removing index-bleed or sample cross-over from datasets is important for downstream community ecology analysis. AMPtk utilizes a mock community as reference point for calculating the rate of index-bleed between samples. It than uses that value to remove read counts from an OTU table that fall below the index-bleed threshold. Each OTU is calculated separately, so that low-abundance OTUs are not indiscriminately removed. More information can be found here.
Usage: amptk filter <arguments>
version: 1.3.0
Description: Script filters OTU table generated from the `amptk cluster` command and should
be run on all datasets to combat barcode-switching or index-bleed (as high as
2%% in MiSeq datasets, ~ 0.3%% in Ion PGM datasets). This script works best when
a spike-in control sequence is used, e.g. Synthetic Mock, although a mock is not required.
Required: -i, --otu_table OTU table
-f, --fasta OTU fasta
Optional: -o, --out Base name for output files. Default: use input basename
-b, --mock_barcode Name of barcode of mock community (Recommended)
-m, --mc Mock community FASTA file. Required if -b passed. [synmock,mock1,mock2,mock3,other]
-c, --calculate Calculate index-bleed options. Default: all [in,all]
-d, --drop Sample(s) to drop from OTU table. (list, separate by space)
--negatives Negative sample names. (list, separate by space)
--ignore Ignore sample(s) during index-bleed calc (list, separate by space)
Filtering -n, --normalize Normalize reads to number of reads per sample [y,n]. Default: y
-p, --index_bleed Filter index bleed between samples (percent). Default: 0.005
-t, --threshold Number to use for establishing read count threshold. Default: max [max,sum,top5,top10,top25]
-s, --subtract Threshold to subtract from all OTUs (any number or auto). Default: 0
--delimiter Delimiter of OTU tables. Default: tsv [csv, tsv]
--min_reads_otu Minimum number of reads for valid OTU from whole experiment. Default: 2
--min_samples_otu Minimum number of samples for valid OTU from whole experiment. Default: 1
--col_order Column order (separate by space). Default: sort naturally
--keep_mock Keep Spike-in mock community. Default: False
--show_stats Show OTU stats on STDOUT
--debug Keep intermediate files.
-u, --usearch USEARCH executable. Default: usearch9
amptk lulu¶
Script runs LULU post-clustering OTU table filtering. see doi:10.1038/s41467-017-01312-x
Usage: amptk lulu <arguments>
version: 1.3.0
Description: Script is a wrapper for the LULU OTU table post-clustering curation of amplicon
data. The script calculates pairwise identity between the OTUs and then filters
the OTU table based on whether closely related OTUs that share the same/similar
distributions in the data are "daughters" of the "parent" OTU. Requires R and the
LULU R package. doi:10.1038/s41467-017-01312-x
Arguments: -i, --otu_table Input OTU table (Required)
-f, --fasta Input OTUs in FASTA format (Required)
-o, --out Output base name. Default: input basename
--min_ratio_type Minimum ratio threshold. Default: min [min,avg]
--min_ratio Minimum ratio. Default: 1
--min_match Minimum match pident (%%). Default: 84
--min_relative_cooccurence Minimum relative co-occurance (%%): Default: 95
--debug Keep intermediate files.
amptk taxonomy¶
This script assigns taxonomy to OTUs and an OTU table. A variety of methods are available, more details are located here.
Usage: amptk taxonomy <arguments>
version: 1.3.0
Description: Script maps OTUs to taxonomy information and can append to an OTU table (optional).
By default the script uses a hybrid approach, e.g. gets taxonomy information from
SINTAX, UTAX, and global alignment hits from the larger UNITE-INSD database, and
then parses results to extract the most taxonomy information that it can at 'trustable'
levels. SINTAX/UTAX results are used if BLAST-like search pct identity is less than 97%%.
If % identity is greater than 97%, the result with most taxonomy levels is retained.
Run amptk info to see taxonomy databases installed.
Arguments: -f, --fasta Input FASTA file (i.e. OTUs from amptk cluster) (Required)
-i, --otu_table Input OTU table file (i.e. otu_table from amptk cluster)
-o, --out Base name for output file. Default: amptk-taxonomy.<method>.txt
-d, --db Select Pre-installed database [ITS1, ITS2, ITS, 16S, LSU, COI]. Default: ITS2
-m, --mapping_file QIIME-like mapping file
-t, --taxonomy Taxonomy calculated elsewhere. 2 Column file.
--method Taxonomy method. Default: hybrid [utax, sintax, usearch, hybrid, rdp, blast]
--add2db Add FASTA files to DB on the fly.
--fasta_db Alternative database of fasta sequenes to use for global alignment.
--utax_db UTAX formatted database. Default: ITS2.udb [See configured DB's below]
--utax_cutoff UTAX confidence value threshold. Default: 0.8 [0 to 0.9]
--usearch_db USEARCH formatted database. Default: USEARCH.udb
--usearch_cutoff USEARCH threshold percent identity. Default 0.7
--sintax_cutoff SINTAX confidence value threshold. Default: 0.8 [0 to 0.9]
-r, --rdp Path to RDP Classifier. Required if --method rdp
--rdp_db RDP Classifer DB set. [fungalits_unite, fungalits_warcup. fungallsu, 16srrna]
--rdp_cutoff RDP Classifer confidence value threshold. Default: 0.8 [0 to 1.0]
--local_blast Local Blast database (full path) Default: NCBI remote nt database
--tax_filter Remove OTUs from OTU table that do not match filter, i.e. Fungi to keep only fungi.
-u, --usearch USEARCH executable. Default: usearch9
--cpus Number of CPUs to use. Default: all
--debug Keep intermediate files
amptk show¶
This utility will count the number of reads for each sample from a demultiplexed FASTQ sample. Additionally it measures read length for the entire dataset and allows you to quality trim using expected errors. Note quality trimming is slow in this script and isn’t intended to be used for normal amplicon dataset processing.
Usage: amptk show <arguments>
version: 1.3.0
Description: Script takes de-multiplexed data (.demux.fq) as input and counts reads per barcode.
Required: -i, --input Input FASTQ file (.demux.fq)
--quality_trim Quality trim reads
-e, --maxee maxEE threshold for quality. Default: 1.0
-l, --length truncation length for trimming: Default: 250
-o, --out Output FASTQ file name (--quality_trim only)
amptk select¶
This script allows you to keep samples from a demultiplexed FASTQ sample, useful for keeping samples that have higher than a --threshold number of reads.
Usage: amptk select <arguments>
version: 1.0.0
Description: Script filters de-multiplexed data (.demux.fq) to select only reads from samples
provided in a text file, one name per line or pass a list to keep to --list.
Required: -i, --input Input FASTQ file (.demux.fq)
-t, --threshold Keep samples with read count greater than -t
-l, --list List of sample (barcode) names to keep, separate by space
-f, --file List of sample (barcode) names to keep in a file, one per line
-o, --out Output file name
--format File format for output file. Default: fastq [fastq, fasta]
amptk remove¶
This script allows you to drop samples from a demultiplexed FASTQ sample, useful for removing samples that have low read counts or are from potentially a different project.
Usage: amptk select <arguments>
version: 1.3.0
Description: Script filters de-multiplexed data (.demux.fq) to select only reads from samples
provided in a text file, one name per line or pass a list to keep to --list.
Required: -i, --input Input FASTQ file (.demux.fq)
-t, --threshold Keep samples with read count greater than -t
-l, --list List of sample (barcode) names to keep, separate by space
-f, --file List of sample (barcode) names to keep in a file, one per line
-o, --out Output file name
--format File format for output file. Default: fastq [fastq, fasta]
amptk sample¶
This script will sub-sample or pseudo-rarefy a dataset to an equal number of reads per sample. Note, this should not be used during standard amplicon community analysis, however, there are some fringe use cases where it is appropriate.
Usage: amptk sample <arguments>
version: 1.3.0
Description: Script sub-samples (rarifies) de-multiplexed data to equal number of reads per
sample. For community analysis, this might not be appropriate as you are ignoring
a portion of your data, however, there might be some applications where it is useful.
Required: -i, --input Input FASTQ file
-n, --num_reads Number of reads to sub-sample to
-o, --out Output FASTQ file name
amptk drop¶
This script allows you to drop OTUs from an OTU table. Usage example would be that you identify OTUs that are from contamination and you want to remove them from the OTU table.
Usage: amptk drop <arguments>
version: 1.3.0
Description: Script drops OTUs from dataset and outputs new OTU table
Required: -i, --input Input OTU file (.cluster.otus.fa) (FASTA)
-r, --reads Demultiplexed reads (.demux.fq) (FASTQ)
-l, --list List of OTU names to remove, separate by space
-f, --file List of OTU names to remove in a file, one per line
-o, --out Output file name. Default: amptk-drop
amptk stats¶
This script is a wrapper for Vegan/Phyloseq and is meant as a first pass overview of your community ecology data. The script takes a BIOM file containing OTU table, taxonomy, and metadata (output of amptk taxonomy). The script than loops through all metadata and returns a hypothesis test (Adonis and Betadisper), an NMDS graph of the data, and an alpha diversity graph. This script requires R, Vegan, and Phyloseq. Script is considered beta as it is new.
Usage: amptk stats <arguments>
version: 1.3.0
Description: A wrapper script for Phyloseq and Vegan R packages that draws NMDS of all
treatments in a BIOM file (output from amptk taxonomy). The script also runs
hypothesis tests (Adonis and Betadispersion) for each treatment.
Arguments: -i, --biom Input BIOM file with taxonomy and metadata (Required)
-t, --tree Phylogeny of OTUs (from amptk taxonomy) (Required)
-d, --distance Distance metric. Default: raupcrick [raupcrick,jaccard,bray,unifrac,wunifrac]
-o, --out Output base name. Default: amptk_stats
--ignore_otus Drop OTUs from table before running stats
Example 1:
amptk stats -i test.biom -t test.tree.phy -o test_stats
-------------------------------------------------------
[Feb 06 09:08 PM]: OS: MacOSX 10.14.3, 8 cores, ~ 17 GB RAM. Python: 3.6.7
[Feb 06 09:08 PM]: R v3.5.1; Phyloseq v1.26.0
[Feb 06 09:08 PM]: Running hypothesis test using bray distance metric on all treatments, drawing NMDS for each.
[Feb 06 09:10 PM]: HTML output files were generated for each treatment: test_stats
-------------------------------------------------------
The script will loop through your treatments and generate an HTML page for each treatment displaying an NMDS ordination, alpha diversity, and some summary stats. Hover-over is enabled on the ordination so you can easily identify outliers.
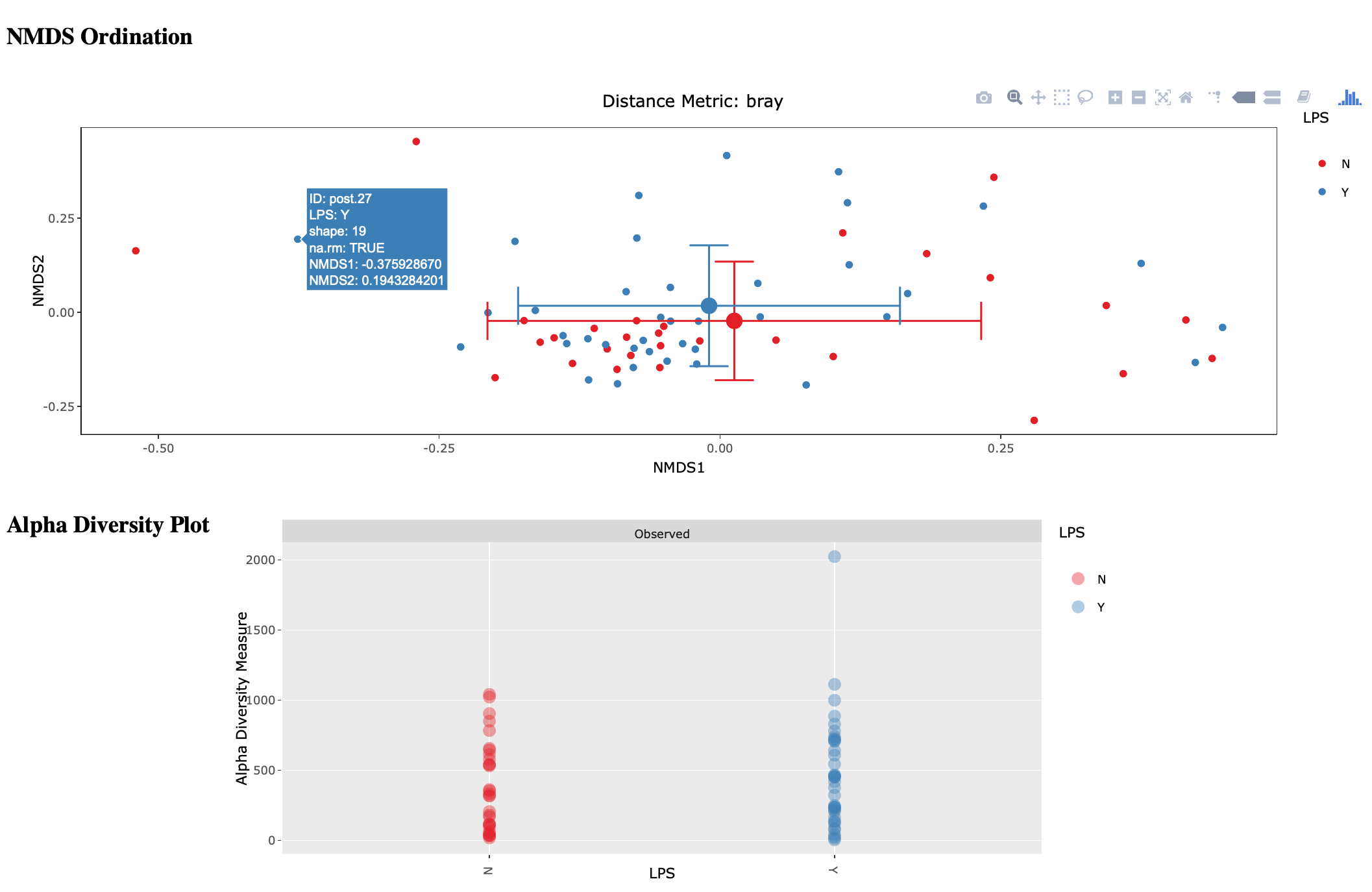
amptk summarize¶
This script will traverse the taxonomy tree from an OTU table that is appended with taxonomy information, i.e. the output of amptk taxonomy. It can optionally produce stacked bar graphs of taxonomy for each level of taxonomy.
Usage: amptk summarize <arguments>
version: 1.3.0
Description: Script traverses the taxonomy information and creates an OTU table for each
level of taxonomy, i.e. Kingdom, Phylum, Class, etc. Optionally, it will
create a Stacked Bar Graph for each taxonomy levels for each sample. Requires
Matplotlib, numpy, and pandas.
Arguments: -i, --table OTU Table containing Taxonomy information (Required)
-o, --out Base name for output files. Default: amptk-summary
--graphs Create stacked Bar Graphs.
--format Image output format. Default: eps [eps, svg, png, pdf]
--percent Convert numbers to Percent for Graphs. Default: off
--font_size Adjust font size for X-axis sample lables. Default: 8
Example 1:
amptk summarize -i test.otu_table.taxonomy.txt --graphs -o test --font_size 6 --format pdf
Example 2:
amptk summarize -i test.otu_table.taxonomy.txt --graphs -o test --font_size 6 --format pdf --percent
amptk funguild¶
FunGuild is a tool for assigning functional information to OTUs. You use this script by simply providing an OTU table that has been appended with taxonomy, i.e. the otu_table.taxonomy.txt from amptk taxonomy.
Usage: amptk funguild <arguments>
version: 1.3.0
Description: Script takes OTU table as input and runs FUNGuild to assing functional annotation to an OTU
based on the Guilds database. Guilds script written by Zewei Song (2015).
Options: -i, --input Input OTU table
-d, --db Database to use [fungi, nematode]. Default: fungi
-o, --out Output file basename.
amptk meta¶
This script is an alternative to using BIOM file format for downstream processing. It takes a metadata file in CSV format with the first column having sample IDs that match sample IDs in an OTU table. The script than pivots the OTU table and appends it to the metadata, which can be imported into something like Vegan in R.
Usage: amptk meta <arguments>
version: 1.3.0
Description: Script takes meta data file in CSV format (e.g. from excel) and an OTU table as input.
The first column of the meta data file must match the OTU table sample headers exactly.
It then pivots the OTU table and appends it to the meta data file.
Required: -i, --input Input OTU table
-m, --meta Meta data table (csv format)
-o, --out Output (meta data + pivotted OTU table)
--split_taxonomy Make separate tables for groups of taxonomy [k,p,c,o,f,g]
amptk heatmap¶
Transform your OTU table into a heatmap using Seaborn and Matplotlib.
Usage: amptk heatmap <arguments>
version: 1.3.0
Description: Script creates a heatmap from an OTU table. Several settings are customizable.
Requires Seaborn, matplotlib, numpy, and pandas.
Arguments: -i, --input Input OTU table (Required)
-o, --output Output file (Required)
-m, --method Type of heatmap. Default: clustermap [clustermap,heatmap]
-d, --delimiter Delimiter of OTU table. Default: tsv [tsv,csv]
-f, --format Figure format. Default: pdf [pdf,jpg,svg,png]
--font Font set. Default: arial
--color Color Palette. Default: gist_gray_r
--figsize Figure size. Default: 2x8
--annotate Annotate heatmap with values.
--distance_metric Distance metric to use for clustermap. Default: braycurtis
--cluster_columns Cluster the columns (samples). Default: False [True,False]
--cluster_method Clustering method for clustermap. Default: single [single,complete,average,weighted]
--scaling Scale the data by row. Default: None [None, z_score, standard]
--yaxis_fontsize Y-Axis Font Size. Default: 6
--xaxis_fontsize X-Axis Font Size. Default: 6
--normalize Normalize data based total, tsv file ID<tab>count
--normalize_counts Value to normalize counts to, i.e. 100000
--vmax Maximum value for heatmap coloration.
--debug Print pandas table on import to terminal
amptk SRA-submit¶
Submitting your data to NCBI SRA can be a real pain, this script tries to make it easier to make that happen. Data submitted to SRA needs to be split up by sample, however it should also be minimally processed -> what I mean by that is that Illumina data should be raw (output of bcl2fastq for example) and 454/Ion Torrent data should be demultiplexed based on sample, but otherwise should not be trimmed. This is where amptk SRA-submit can help. The script takes the raw input and outputs gzipped FASTQ files that are minimally processed for SRA. Moreover, if you create a BioProject and BioSamples for each of your samples prior to running the script, you can bass the BioSample worksheet from NCBI to the script and it will automatically generate an SRA submission file. You can customize some of the text in that file, i.e. via the --description argument.
Usage: amptk SRA-submit <arguments>
version: 1.3.0
Description: Script aids in submitted your data to NCBI Sequence Read Archive (SRA) by splitting
FASTQ file from Ion, 454, or Illumina by barcode sequence into separate files for
submission to SRA. This ensures your data is minimally processed as only barcodes
are removed. However, you can assert that primers must be found in order for
sequences to be processed. Additionally, you can pass the --biosample argument
with an NCBI biosample tab-delimited file and the script will auto-populate an
SRA submission file.
Arguments: -i, --input Input FASTQ file or folder (Required)
-o, --out Output base name. Default: sra
-m, --mapping_file QIIME-like mapping file.
-b, --barcode_fasta Mulit-fasta file containing barcodes used.
-s, --biosample BioSample worksheet from NCBI (from confirmation email)
-p, --platform Sequencing platform. Defalt: ion (ion, illumina, 454)
-n, --names CSV name mapping file, e.g. BC_1,NewName
-d, --description Paragraph description for SRA experimental design. Use quotes to wrap paragraph.
-f, --fwd_primer Forward primer sequence. Default: fITS7
-r, --rev_primer Reverse primer sequence. Default: ITS4
-a, --append Append a name to the output of all files in run, i.e. run1 -> Sample_run1
--primer_mismatch Number of mismatches allowed in primer search. Default: 2
--barcode_mismatch Number of mismatches in barcode to allow. Default: 0
--require_primer Require primer(s) to be present for output. Default: off [off,forward,both]
--min_len Minimum length read to keep after trimming barcodes. Default 50
---force Overwrite directory with same name
AMPtk Setup¶
amptk install¶
This simple script will download and unpack the pre-build reference databases.
Usage: amptk install <arguments>
version: 1.3.0
Description: Script downloads pre-formated databases for use with the `amptk taxonomy`
command. You can download databases for fungal ITS, bacterial 16S, fungal
LSU, or arthropod/chordate COI amplicons.
Arguments: -i Install Databases. Choices: ITS, 16S, LSU, COI
--force Over-write existing databases
amptk database¶
Building reference databases is done with amptk database. It has built-in parsers for UNITE and RDP FASTA headers, see the discussion about AMPtk taxonomy <taxonomy> for more information on FASTA headers.
Usage: amptk database <arguments>
version: 1.3.0
Description: Setup/Format reference database for amptk taxonomy command.
Arguments: -i, --fasta Input FASTA file
-o, --out Base name for output files, i.e. ITS2
-f, --fwd_primer Forward primer. Default: fITS7
-r, --rev_primer Reverse primer. Default: ITS4
--format Reformat FASTA headers to UTAX format. Default: unite2utax [unite2utax, rdp2utax, off]
--drop_ns Removal sequences that have > x N's. Default: 8
--create_db Create a DB. Default: usearch [utax, usearch]
--skip_trimming Keep full length sequences. Default: off
--derep_fulllength Remove identical sequences.
--lca Run LCA (last common ancestor) on taxonomy if dereplicating sequences.
--min_len Minimum length to keep. Default: 100
--max_len Maximum length to keep. Default: 1200
--trunclen Truncate records to length.
--subsample Random subsample reads.
--primer_mismatch Max Primer Mismatch. Default: 2
--keep_all Keep Sequence if forward primer not found.
--utax_trainlevels UTAX custom parameters. Default: kpcofgs
--utax_splitlevels UTAX custom parameters. Default: NVkpcofgs
--cpus Number of CPUs to use. Default: all
--install Install into AMPtk Database
-u, --usearch USEARCH executable. Default: usearch9
amptk primers¶
This command lists the primers that are available via their names. You can always input the actual primer sequence.
----------------------------------
Primers hard-coded into AMPtk:
----------------------------------
16S_V3 CCTACGGGNGGCWGCAG
16S_V4 GACTACHVGGGTATCTAATCC
515FB GTGYCAGCMGCCGCGGTAA
806RB GGACTACNVGGGTWTCTAAT
COI-F GGTCAACAAATCATAAAGATATTGG
COI-R GGWACTAATCAATTTCCAAATCC
ITS1 TCCGTAGGTGAACCTGCGG
ITS1-F CTTGGTCATTTAGAGGAAGTAA
ITS2 GCTGCGTTCTTCATCGATGC
ITS3 GCATCGATGAAGAACGCAGC
ITS3_KYO2 GATGAAGAACGYAGYRAA
ITS4 TCCTCCGCTTATTGATATGC
ITS4-B CAGGAGACTTGTACACGGTCCAG
ITS4-B21 CAGGAGACTTGTACACGGTCC
JH-LS-369rc CTTCCCTTTCAACAATTTCAC
LR0R ACCCGCTGAACTTAAGC
LR2R AAGAACTTTGAAAAGAG
fITS7 GTGARTCATCGAATCTTTG